SYNGAP1, un gen clave para el desarrollo cerebral y la cognición
Hoy en Dciencia tenemos un artículo de divulgación sobre una enfermedad poco frecuente. Se trata de un artículo de divulgación científica, escrito por Gemma Gou Alsina, investigadora del grupo del Dr. Àlex Bayés, del Hospital de la Sant Creu i Sant Pau de Barcelona, pero antes pasar a la parte científica os dejamos con una pequeña introducción de un padre de una niña que sufre esta alteración genética. Esperamos que os guste y os interese.
Cuando nos confirman que vamos a ser padres, parece que damos por hecho que todo siempre va a ir bien, y no somos conscientes de que una pequeña mutación en un único gen es capaz de ponerte el mundo patas arriba.
Hace 3 años, cuando recibimos el diagnóstico, solo había 2 familias en España y 300 en el mundo con esta enfermedad, ahora ya somos 20 en España y más de 500 en total.
Necesitamos dar visibilidad al SYNGAP1 porque estamos convencidos de que muchísimos niños que quedan en el olvido de las ‘enfermedades raras de origen desconocido’, pueden tener su diagnóstico aquí, en este gen.
Nosotros, las familias, hacemos cuanto podemos por difundir este mensaje, pero necesitamos de vosotros, la comunidad científica, para cerrar el círculo. Los investigadores, biólogos, pediatras, neurólogos, etc. tienen que saber qué es el SYNGAP1 y qué consecuencias tiene en nuestros hijos, para poder identificarlo cuanto antes, solicitar las pruebas oportunas y ayudar a los padres y madres desde el principio.
Por eso, gracias por permitirnos usar vuestro altavoz y gracias por aprender un poco más acerca de este gen.
Gonzalo Bermejo
Padre de una niña afectada por SYNGAP1 y autor del blog www.elblogdegonzalobermejo.wordpress.com
Presidente de ‘Syngap1 España’
SYNGAP1, un gen clave para el desarrollo cerebral y la cognición
Pensar cómo resolver un problema, aprender que el fuego puede hacernos daño, memorizar la lista de la compra o tomar una decisión son ejemplos de procesos cognitivos que requieren de la correcta ejecución de nuestro cerebro. Para ello es necesario que más de 100 billones de neuronas se comuniquen adecuadamente mediante “puentes” llamados sinapsis, los cuales permitirán la formación de complejas redes neuronales (Fig. 1). En esta comunicación, una neurona presináptica (o emisora) va a liberar un mensaje particular, normalmente a través de su axón, usando códigos (compuestos químicos) que suelen ser neurotransmisores. A través de la sinapsis (canal de comunicación), la neurona postsináptica (receptora de la información) mediante sus dendritas va a recibir y descodificar el mensaje liberado en el espacio entre ambas neuronas con la ayuda de diferentes moléculas (Fig. 1). Cuando se envía un mensaje que va a favorecer la activación neuronal, hablamos de sinapsis excitatoria o si produce el efecto contrario, de sinapsis inhibitoria.
Se estima que en el cerebro más de un 90% de las sinapsis son excitatorias y mediadas por el neurotransmisor glutamato. La mayoría de estas sinapsis glutamatérgicas cuentan con protuberancias a modo de “antenas receptoras” localizadas en las dendritas de las neuronas postsinápticas llamadas espinas dendríticas. En estas espinas se encuentra una especie de matriz denominada densidad postsináptica que podría llegar a albergar hasta 2.000 proteínas (Fig. 1). Esta estructura es la encargada de descodificar el mensaje recibido por la neurona presináptica y transmitir la información apropiada a la neurona postsináptica. Anomalías en alguno de sus componentes, ya sea a nivel de abundancia o función, están asociadas a varias enfermedades neuropsiquiátricas como la discapacidad intelectual o el trastorno del espectro autista.

Figura 1. La sinapsis glutamatérgica y sus componentes. Composición de diagramas modificados de Dietrich et al. 2016 y Kennedy et al. 2016. La imagen del cerebro ha sido extraída de www.turbosquid.com
El gen SYNGAP1 y la proteína SynGAP
Imaginemos que cada célula de nuestro cuerpo contiene una inmensa biblioteca dónde cada estantería sería lo que conocemos como cromosoma y cada uno de sus libros, un gen. Las copias de un gen son lo que llamamos transcritos y darán las instrucciones para generar diferentes proteínas con funciones concretas (para más información ver artículo de DCiencia “ADN, genes, cromosomas…”). En particular, el gen SYNGAP1 se encuentra en el cromosoma autosómico 6p21.32 en humanos y codifica para la proteína SynGAP, de las siglas “Synaptic GTPase Activating Protein”. Se trata de un gen complejo ya que puede dar lugar a distintos transcritos que a su vez se traducirán a diferentes isoformas de SynGAP, es decir proteínas con una parte común a todas ellas, pero con ligeras variaciones en las secuencias de sus extremos. La proteína SynGAP fue descubierta el 1998 por dos laboratorios independientes de Estados Unidos de América y años después se estimó que podrían identificarse por lo menos 12 isoformas en mamíferos. Se expresa desde fases embrionarias y principalmente en el cerebro. Inicialmente se caracterizó como una de las proteínas más abundantes de la densidad postsináptica, aunque también tendría una localización y función no sináptica preponderante en etapas iniciales del neurodesarrollo. SynGAP, además de cumplir un rol estructural por su tamaño y abundancia en la sinapsis, una vez activada, ejercería como un semáforo en rojo o inhibidor de las cascadas de señalización iniciadas por sus moléculas diana (por ej. HRas, Rap1/2 o Rab5).
En general, SynGAP regula distintos tipos de plasticidad sináptica tanto a nivel estructural (por ej. regulando su morfología y composición de receptores de glutamato) como funcional (por ej. modulando procesos de potenciación o inhibición a corto y largo plazo), eventos relacionados con la correcta conectividad neuronal y el balance excitatorio/inhibitorio. De hecho, ratones modificados genéticamente para que no expresen por completo Syngap1, mueren durante la primera semana de vida, indicando la importancia de SynGAP ya en edades muy tempranas del desarrollo. A pesar de que en los últimos 20 años se ha avanzado mucho en el conocimiento de esta proteína, aún quedan muchas preguntas por responder ya que su neurobiología es mucho más compleja de lo que previamente se anticipó. Por ejemplo, se ha visto que las distintas isoformas de SynGAP muestran un patrón de expresión y distribución espacio-temporal particular, una regulación diferencial de las mismas proteínas diana o incluso papeles opuestos en la sinapsis.
La haploinsuficiencia de SYNGAP1 causa discapacidad intelectual
Cuando una de las dos copias de SYNGAP1 (ya sea paterna o materna) alberga una mutación con pérdida de función (imagínese que le faltan páginas al libro y no se puede copiar entero), dará lugar a una haploinsuficiencia de SYNGAP1. Consecuentemente, la neurona no llegará a expresar los niveles necesarios de proteína SynGAP (sólo se expresaría el ~50%) dando lugar a discapacidad intelectual moderada o severa con alta afectación en el lenguaje, de epilepsia generalizada (en > 80% de los pacientes), trastorno del espectro autista (en el 50% de los casos) y un retraso en el desarrollo psicomotor global evidente ya a los 2-3 años de edad. Además, alrededor de un 75% de los casos padecen problemas de conducta tales como hiperexcitabilidad, comportamiento agresivo, autolesión o déficit de conducta innata de supervivencia. En algunos pacientes también se han visto alteraciones en el procesamiento sensorial (por ej. estímulos relacionados con el tacto, el dolor o sincronización sensorial), trastornos musculoesqueléticos (por ej. ataxia o hipotonía), microcefalia, convulsiones, trastorno del sueño, estrabismo, estreñimiento, dificultades en la ingesta o rasgos dismórficos faciales (Fig. 2). También es interesante destacar que SYNGAP1 se considera un gen de riesgo para otros trastornos del neurodesarrollo como la esquizofrenia, trastorno bipolar o el déficit de atención con hiperactividad.
La patología asociada a SYNGAP1 se identificó por primera vez en Canadá el 2009 por el Dr. Michaud y sus colaboradores (Hamdan et al. 2009). Hasta la fecha, solo se ha descrito un caso de herencia parental debido a un mosaicismo somático (cuando algunas células son normales mientras que otras llevan la mutación). Por lo tanto, en el resto de los casos descritos, la enfermedad sería causada por una mutación espontánea (de novo) de SYNGAP1. Este hecho implica que el riesgo de que hermanos de esas personas afectadas padezcan la enfermedad sea bajo y afecte por igual en ambos sexos.
En 2018 se estimó que la haploinsuficiencia de SYNGAP1 en la población tendría una incidencia de 1-4/10.000 individuos, y que representa del 0,5 al 1% de todos los casos de discapacidad intelectual en el mundo, frecuencia similar a la descrita para el síndrome de frágil X. Sin embargo, en los últimos años el número de afectados no ha parado de crecer. Para ilustrarlo, en 2018 se habían registrado alrededor de 200 casos a nivel mundial mientras que actualmente ya son más de 500 casos de los cuales 20 se encuentran en España. Además, dado que no se sabe si la esperanza de vida de estos pacientes se ve afectada y se ha descrito una paciente de 31 años de edad (Prchalova et al. 2017), seguramente podría haber muchos más casos entre las personas con discapacidad intelectual sin diagnóstico de las descritas en la actualidad.
Tradicionalmente se han considerado las mutaciones que llevan a la haploinsuficiencia de SYNGAP1 como causantes de discapacidad intelectual no sindrómica, por la falta (en algunos casos) de anomalías físicas evidentes que faciliten el diagnóstico, o de retraso mental de tipo 5 (OMIM: #603384). No obstante, en 2015 se introdujo el concepto de síndrome asociado a SYNGAP1 (Parker et al. 2015; Fig. 2) mientras que posteriormente otros científicos establecieron la terminología de encefalopatía epiléptica de SYNGAP1 (Vlaskamp y Scheffer 2019) o de encefalopatía del desarrollo y epiléptica asociada a SYNGAP1 (Orphanet: #544254).
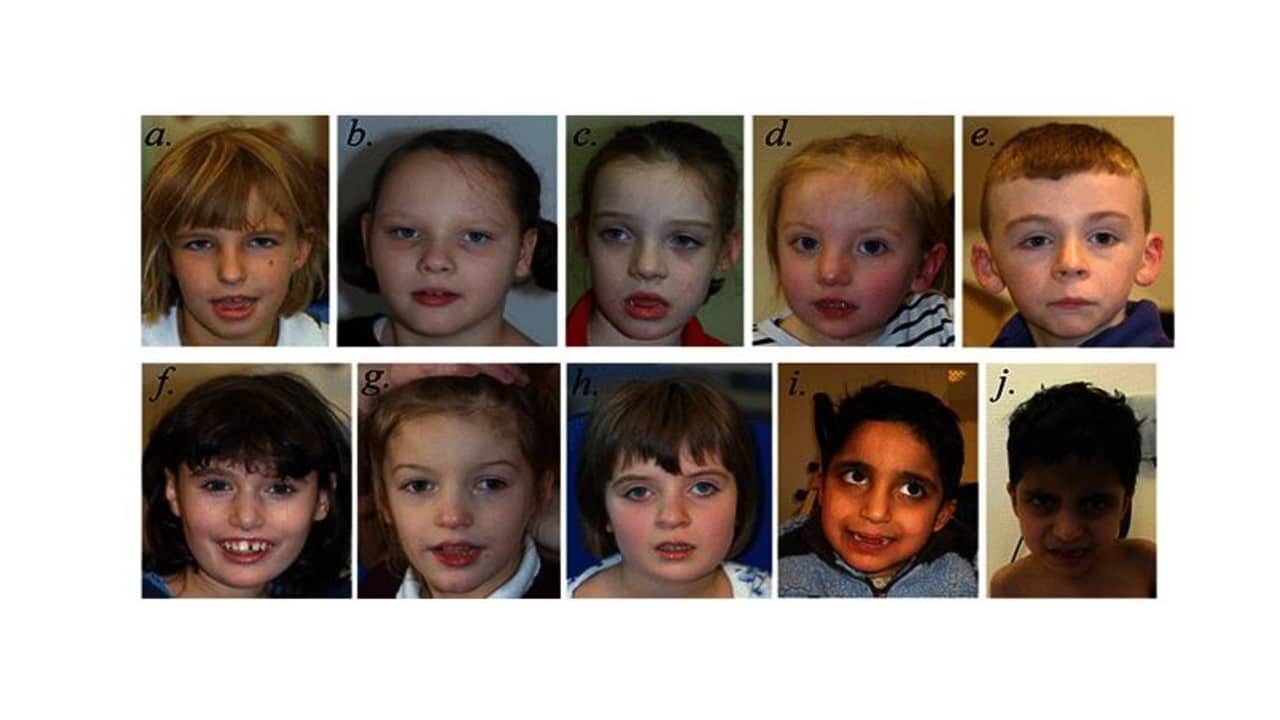
Figura 2. Rasgos faciales de individuos con haploinsuficiencia de SYNGAP1. Estudio realizado e imágenes extraídas de Parker et al. 2015.
Diagnóstico y tratamiento de pacientes haploinsuficientes para SYNGAP1
En la actualidad, no hay un criterio de diagnóstico establecido. No obstante, cuando un niño presenta retraso en el desarrollo o discapacidad intelectual con o sin epilepsia generalizada y/o trastorno del espectro autista, se recomienda llevar a cabo una prueba genética. Esta prueba típicamente consiste en detectar desbalances cromosómicos o si hay alteraciones en genes candidatos causantes de discapacidad intelectual (dónde debería incluirse SYNGAP1) mediante un panel multigénico. Alternativamente, se puede llevar a cabo una secuenciación del exoma o del gen en concreto. Todas estas pruebas son las que permitirán identificar si una de las copias de SYNGAP1 es patógena y es la causa del cuadro clínico que presenta el paciente evaluado.
Varios estudios liderados por el Dr. Rumbaugh del The Scripps Research Institute (Florida, EUA) demostraron por primera vez en ratones que existiría un período crítico del neurodesarrollo (equivalente a los primeros años de vida en humanos) en el cual la falta de los niveles adecuados de SynGAP resultaría en una maduración prematura de las sinapsis y la reducción de sus niveles de plasticidad, dando lugar a los defectos cognitivos identificados en los individuos afectados. Además, se postuló que, una vez superado este período crítico, las alteraciones derivadas de la falta de SynGAP serían irreparables. Sin embargo, recientemente se ha visto que la recuperación de la expresión total de esta proteína en la adultez también revertiría algunas de las alteraciones. Por todo ello, sería ideal identificar la haploinsuficiencia de SYNAGAP1 lo antes posible y conseguir que las neuronas expresaran los niveles adecuados de proteína, especialmente durante aquellos períodos críticos relacionados con la adquisición de habilidades y la expresión de determinadas conductas.
Desafortunadamente no hay un tratamiento efectivo para los pacientes afectados por una haploinsuficiencia de SYNGAP1. El tratamiento actual administrado se basa en terapia combinada e individualizada para corregir los diferentes síntomas (para más información ver: https://www.ncbi.nlm.nih.gov/books/NBK537721/), ya que no todos los pacientes presentan exactamente el mismo cuadro clínico ni responden igual a los tratamientos (por ej. hay niños fármaco-resistentes a determinados antiepilépticos mientras otros no lo son). En cuanto a la investigación científica, son pocos los estudios que han descrito los efectos de posibles fármacos o la capacidad de curar esta enfermedad mediante terapia génica ya sea en modelos animales de la patología o en humanos. Solo algunos de los fármacos testados entre los cuales se incluyen inhibidores de la vía de señalización MEK-ERK (se encuentra hiperactivada en estos modelos animales), estatinas (Lovastatina o Rosuvastatin) o antiepilépticos como el Parampanel han conseguido atenuar algunos síntomas.
Apuntes finales
Si se sospecha que algún familiar o conocido pueda padecer una haploinsuficiencia de SYNGAP1 o quieren contribuir a la mejora de la calidad de vida de estas personas, pueden ponerse en contacto con la asociación española de SYNGAP1 (www.syngap1españa.es). A través de esta plataforma podrán compartir y aprender de la experiencia de otras familias afectadas, así como sumarse a los retos ya sean económicos o sociales que proponen.
Bibliografía
-Cook et al. Lovastatin Treatment of a Patient with a De Novo SYNGAP1 Protein Truncating Variant. 2019. J Child Adolesc Psychopharmacol. May;29(4):321-322. doi: 10.1089/cap.2018.0159.
-Creson and Rojas et al. Re-expression of SynGAP protein in adulthood improves translatable measures of brain function and behavior. Elife. 2019 Apr 26;8. pii: e46752. doi: 10.7554/eLife.46752. e14988. doi: 10.1111/jnc.14988.
-Dieterich, Daniela C., and Michael R. Kreutz. 2016. ‘Proteomics of the Synapse – A Quantitative Approach toNeuronal Plasticity’. Molecular & Cellular Proteomics 15 (2): 368. https://doi.org/10.1074/mcp.R115.051482.
-Gamache et al. Twenty Years of SynGAP Research: From Synapses to Cognition. J Neurosci. 2020. Feb19;40(8):1596-1605. doi: 10.1523/JNEUROSCI.0420-19.2020.
-Gou et al. SynGAP Splice Variants Display Heterogeneous Spatio-Temporal Expression And Subcellular DistributionIn The Developing Mammalian Brain. 2020. J Neurochem.
-Hamdan et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 2009. Feb 5;360(6):599-605. doi: 10.1056/NEJMoa0805392.
-Jeyabalan y Clement. SYNGAP1: Mind the Gap. 2016. Front Cell Neurosci. 10:32. doi: 10.3389/fncel.2016.00032.
-Kennedy, Mary B. 1993. ‘The Postsynaptic Density’. Current Opinion in Neurobiology 3 (5): 732–37. https://doi.org/10.1016/0959-4388(93)90145-O.
-Kilinc et al. Species-conserved SYNGAP1 phenotypes associated with neurodevelopmental disorders. 2018. Mol CellNeurosci. 91:140-150. doi: 10.1016/j.mcn.2018.03.008.
-Kluger et al. Positive Short-Term Effect of Low-Dose Rosuvastatin in a Patient with SYNGAP1-Associated Epilepsy.2019. Neuropediatrics. 50(4):266-267. doi: 10.1055/s-0039-1681066.
-Kopanitsa et al. Chronic treatment with a MEK inhibitor reverses enhanced excitatory field potentials in Syngap1+/- mice. 2018. Pharmacol Rep. 70(4):777-783. doi: 10.1016/j.pharep.2018.02.021.
-Parker et al. De novo, heterozygous, loss‐of‐function mutations in SYNGAP1 cause a syndromic form of intelectual disability. 2015. SAm J Med Genet A. 2015 Oct;167A(10):2231-7. doi: 10.1002/ajmg.a.37189.
-Prchalova et al. Analysis of 31-year-old patient with SYNGAP1 gene defect points to importance of variants in broader splice regions and reveals developmental trajectory of SYNGAP1-associated phenotype: case report. 2017. BMC Med Genet. 18(1):62. doi: 10.1186/s12881-017-0425-4.
-Sullivan et al. Low-Dose Perampanel Rescues Cortical Gamma Dysregulation Associated With Parvalbumin Interneuron GluA2 Upregulation in Epileptic Syngap1+/- Mice. Biol Psychiatry. 2020 Jan 10. pii: S0006-3223(20)30002-0. doi: 10.1016/j.biopsych.2019.12.025.
-Vlaskamp et al. SYNGAP1 encephalopathy: A distinctive generalized developmental and epileptic encephalopathy. 2019 Neurology. Jan 8;92(2):e96-e107. doi: 10.1212/WNL.0000000000006729.
-Weldon et al. The first international conference on SYNGAP1-related brain disorders: a stakeholder meeting of families, researchers, clinicians, and regulators. 2018. J Neurodev Disord. 10(1):6. doi: 10.1186/s11689-018-9225-1


About the Author: Gemma Gou Alsina
2 Comments
Leave A Comment Cancelar la respuesta
Este sitio usa Akismet para reducir el spam. Aprende cómo se procesan los datos de tus comentarios.






Es increíble como el avance del estudio del origen de algunos rasgos pensados antes como enfermedades individuales, ahora se descubren que tienen un origen y desarrollo más complejo, cómo influye en este caso la deficiencia del syngap1 en los primeros años de vida, donde se demarca con más fuerza los efectos de su deficiencia, para posteriormente en su adultez recuperar algunas de estas funciones corporales antes no definidas. Es bueno saber que existe desde el año 2009 bases para aminorar el padecimiento de las personas que tienen esta patología.
la haploinsuficiencia de este gen SYNGAP1 causa estrabismo, una condición ocular que pueden conocer más visitando https://arisvisionmexico.com/padecimientos/padecimiento-estrabismo.html gracias por compartir esta información