Medicina hiperpersonalizada: medicamentos para un paciente único. El milagro de Mila
Hace no demasiados días salió en varios medios una noticia que a lo mejor os ha pasado desapercibida. Se trata de la creación del primer fármaco totalmente personalizado, para un único paciente. Vamos a intentar explicaros en qué ha consistido este caso.
LA ENFERMEDAD
La enfermedad de Batten es el nombre que se emplea para denominar a un grupo de desórdenes del sistema nervioso. El nombre técnico es lipofuscinosis neuronal ceroidea o CLN.
¿Qué es lo que falla en esta enfermedad?
Es una enfermedad genética hereditaria provocada por una mutación en un solo gen. Para cada gen tenemos dos copias (llamadas alelos), una que viene de nuestra madre y la otra de nuestro padre. La mayoría de las CLNs se heredan de forma autosómica recesiva, es decir la mutación se tiene que producir a la vez en las dos copias de ADN para que se produzca la enfermedad . Hasta la fecha se han identificado diez tipos genéticos diferentes de enfermedad de Batten, denominados de CLN1 a CLN10.
Cuando esta mutación recesiva ocurre provoca fallos en el interior de la célula, concretamente en unos orgánulos que se llaman lisosomas. Los lisosomas son como una especie de saquitosunidos por dentro a la membrana de la célula. Dentro de ellos hay unas proteínas con actividad enzimática que son capaces de hidrolizar, romper, macromoléculas para convertirlas en otras mucho más pequeñas. Entre otras cosas, los lisosomas se encargan de reciclar los restos celulares que ya no se van a utilizar. También pueden destruir restos de virus y bacterias. Es decir, los lisosomas actúan como plantas de reciclaje intracelular. Si queréis saber más sobre ellos, podéis ver este vídeo:
Bien, pues en los pacientes que tienen alguna de las variantes de la enfermedad de Batten, sus células no son capaces de reciclar algunas moléculas, que se van acumulando en el interior de las neuronas. También se acumula lipofuscina, que es una sustancia de color amarillo, formada por grasas y proteínas que se produce de manera natural cuando los lisosomas degradan lípidos. Estos acúmulos de, podríamos llamar, basura intracelular, provoca que las células no funcionen bien y aparezcan los síntomas de la enfermedad.
¿Cómo se manifiesta la enfermedad?
La mayor parte de las CLNs empiezan a manifestarse en la infancia. Los síntomas más comunes incluyen pérdida de visión, movimientos anormales (ataxia), dificultades de concentración, pérdida de las capacidades ya adquiridas, demencia. Generalmente se trata de pacientes cuyo estado físico va degenerando y acaban en silla de ruedas, encamados, sin capacidad de comunicarse. Su esperanza de vida es pequeña, aunque depende mucho del tipo de enfermedad de Batten del que se trate. Si los síntomas se manifiestan temprano, generalmente el paciente no supera la infancia, mientras que si son más tardíos, puede llegar a la adolescencia o incluso a los treinta. En los casos en los que la enfermedad se desarrolla en la etapa adulta, no afecta significativamente a la esperanza de vida.
No existe tratamiento para esta enfermedad. Para el tipo 2 la FDA aprobó en 2017 una terapia llamada cerliponasa alfa (Brineura®), que aunque no cura, disminuye o detiene los síntomas.
LA PACIENTE
Mila Makovec tiene ocho años. Hasta los tres parecía una niña sana, sin ningún problema de salud importante. A esa edad, los padres se dieron cuenta de que ese quedaba atascada con algunas palabras y de que un pie se le torcía un poco hacia dentro. A los cuatro años empezó a pegarse los libros a la cara para poder verlos. A los cinco a veces se caía de espaldas y hacía movimientos extraños con los pies. Pocos años después se ha quedado ciega, no puede ponerse de pie ni levantar la cabeza. Además, se tiene que alimentar por medio de una sonda de alimentación y sufre frecuentes convulsiones cada día (hasta treinta). En 2016, con seis años, le diagnosticaron la enfermedad de Batten, concretamente el tipo CLN7.
¿DÓNDE ESTÁ LA ALTERACIÓN MOLECULAR DE MILA?
En diciembre de 2016 los médicos que trataban a Mila en Colorado decidieron secuenciar la parte codificante del genoma de Mila (los llamados exones, la parte del ADN que da lugar a proteínas) para intentar encontrar la causa de su enfermedad. El análisis de ADN logró identificar una mutación en el gen denominado CLN7. Este gen lleva la información para que se sintetice una proteína que participa en el movimiento de las moléculas a través de la membrana de los lisosomas. Lo extraño era que la mutación se encontraba solo en una de las dos copias de su ADN. Solo la copia paterna presentaba la mutación. ¿Y entonces, por qué Mila había desarrollado la enfermedad? Para comprender mejor qué pasaba era necesario secuenciar el genoma completo de Mila, es decir, no solo la parte codificante (exones) sino también la parte no codificante (los llamados intrones). Se trata de una prueba cara y de difícil interpretación. A principios de 2017 el caso llegó al neurólogo Timothy Yu, del Boston Children’s Hospital Su mujer, médico también, leyó el caso en un grupo de Facebook y decidieron ayudar. En solo un mes, el laboratorio de Yu tenía los datos. Efectivamente confirmaron los datos del análisis de los médicos de Colorado, pero se dieron cuenta también de que la parte no codificante de la copia materna del gen CLN7 no correspondía a la secuencia normal: ¡había una secuencia de unos 2.000 nucleótidos que no debería estar ahí! Se trataba de un transposón. Este nombre tan raro solo significa que es un fragmento de ADN que es capaz de copiarse e integrarse y aparecer en cualquier parte del ADN (proceso llamado transposición). En concreto era un retrotransposón, un fragmento de ADN que para moverse utiliza un intermediario de ARN. Este retrotransposón provocaba un error cuando el alelo CLN7 de la madre se debía copiar a ARN mensajero, para producir la proteína. El transposon funciona como un exón extra y produce un splicing incorrecto, pero también se produce ARNm correcto. Como consecuencia, se producía una proteína más corta, que no funcionaba. Por lo tanto, Mila tenía “mal” las dos copias de su gen CLN7, aunque cada copia tenía una alteración distinta.
¿CÓMO “ARREGLAR” ESTE FALLO MOLECULAR?
El equipo del doctor Yu decidió intentar corregir el error en la copia de la madre (provocado por la incorporación del retrotransposón). Se optó por un abordaje terapéutico que consistía en la utilización de oligonucleótidos antisentido (ASOs). Esto puede sonar muy raro, pero es fácil de entender. Vamos a explicarlo un poquito. Un oligonucleótido antisentido es simplemente una cadena de 13 a 25 nucleótidos que es capaz de unirse a un ARN mensajero (ya que es complementario a él). Al unirse a este ARN mensajero lo “bloquea”, de tal manera que no se puede generar la proteína. En el caso de Mila, el fármaco bloquea el ARNm incorrecto, pero permite el ARNm correcto que viene de la madre.
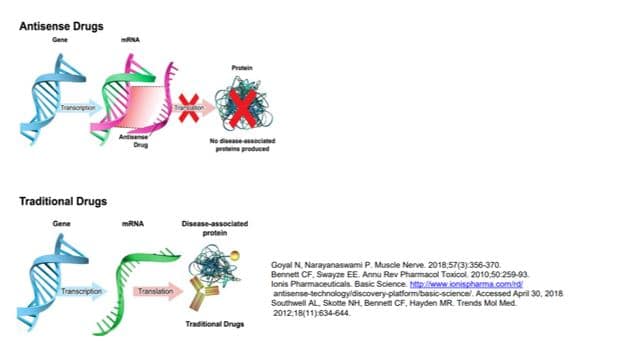
El equipo de Yu diseñó el oligonucleótido antisentido y comprobó que efectivamente funcionaba en células de la piel extraídas de la niña y mantenidas en cultivo. Inmediatamente después encontró una empresa que lo podía fabricar rápidamente. En diciembre de 2017, la nueva terapia ya estaba disponible y se le puso el nombre de milasen. La FDA actuó rápidamente y enero de 2018 concedió la aprobación para el tratamiento.
Aquí tenéis un vídeo elaborado por el propio Boston Children’s Hospital para explicar el caso de Mila:
El tratamiento comenzó con una infusión en la médula espinal de dosis bajas de milasen. Posteriormente la dosis se fue aumentando cada dos semanas, cuando veían que era bien tolerado por Mila.
RESULTADOS
Los efectos en Mila han sido positivos. Tras un año de tratamiento, aunque no se puede hablar de curación, sí de mejora en algunos aspectos., Las convulsiones han disminuido muchísimo, las puntuaciones obtenidas en las pruebas neurológicas se han estabilizado o han mejorado y su volumen cerebral está disminuyendo. No solo eso, sino que se ven algunas otras mejorías, como una ganancia de fuerza en el torso y en las piernas, mayor estado de alerta, mayor capacidad para tragar… Mila está clínicamente estable y no empeora. Sin embargo, sigue sin poder hablar ni ver y necesita ayuda para caminar.
No se ha encontrado una “cura mágica”, pero sí se ha logrado al menos una opción para pacientes que hasta ahora no tenían ninguna, que solo podían ver como día a día sus capacidades vitales se iban degradando más y más hasta finalmente morir.
MEDICINA HIPERPERSONALIZADA
Tras este caso, una de las cuestiones que se debaten es si la llamada medicina hiperpersonalizada, en la que se diseñan y fabrican medicamentos específicos para pacientes concretos, podrá ser una realidad o no, teniendo en cuenta que hablamos de costes de millones de dólares en cada caso. Pese a que no ha trascendido el coste del proceso completo del tratamiento de Mila, se estima que ronda entre los 3 y 5 millones de dólares. El tratamiento se costeó con fondos del propio Boston Children’s Hospital, becas de investigación y dos fundaciones privadas, incluyendo la Fundación creada por los padres de Mila, la Mila’s Miracle Foundation.

El propio doctor Yu ha afirmado que ya más de cien padres se han puesto en contacto con él para pedirle ayuda para el tratamiento de sus hijos. Obviamente es imposible que su equipo ayude a todos, aunque ha comenzado un proyecto piloto para lograr tratar de manera totalmente personalizada a doce pacientes.
Por otra parte, la FDA ha expresado su preocupación o sus dudas en este tipo de casos que ellos llaman ensayos de n=1, es decir, ensayos clínicos en los que solo hay un paciente. Está intentando desarrollar una serie de criterios para que sean seguidos por los médicos en este tipo de situaciones.
Fuentes:
Sobre la enfermedad: https://www.ninds.nih.gov/disorders/patient-caregiver-education/fact-sheets/batten-disease-fact-sheet
El artículo: https://www.nejm.org/doi/full/10.1056/NEJMoa1813279
Fundación Mila’s Miracle: https://www.stopbatten.org/


About the Author: Alberto Morán
14 Comments
Leave A Comment Cancelar la respuesta
Este sitio usa Akismet para reducir el spam. Aprende cómo se procesan los datos de tus comentarios.






La investigación permite mutar las alteraciones genéticas mutantes aleatoriamente. Maravilloso.
Este artículo parécenos moi interesante e necesario. Trata dun tema mi importante, como a creación de medicamentos específicos para a curación de enfermedades pouco comúns. Oxalá que esta investigación siga crecendo e formándose para así axudar a máis personas e reciba máis medios necesarios para seguir profundizando.
Gracias por el comentario
Gracias por el trabajo realizado, esperemos que dispongan de medios para continuar con esta labor.
Gran trabajo de la medicina actual, esperemos que se pueda acabar curando
Gran trabajo de los médicos y un artículo buenísimo
Muchas gracias. Un saludo
Este artículo me ha parecido súper interesante, lo que puedo resaltar es la manera en la que se explican las cosas, cada uno de los puntos que se van tratando. El tema es muy atrayente pues nunca he escuchado hablar sobre la medicina hiperpersonalizada, y mucho menos de la enfermedad de Batten, lo recalco, es un excelente contenido que me deja muy satisfecha por su detallada explicación. Felicitaciones!
Este articulo desde mi punto de vista me parece muy interesante pero a la vez intrigante ya que trata de un tema hasta ahora un poco desconocido como es la creación de un medicamento personalizado, es decir hecha específicamente para una sola persona con el fin de curar ciertas enfermedades no tan comunes. En la actualidad se están realizando varias investigaciones para determinar si va a ser beneficiosa o no.Ademas me parece que esta es una gran opción para ayudar a las personas a lograr la enfermedad que le hayan diagnosticado, como es el caso de Mila que luego de un año de tratamiento ha logrado mejorar en ciertos aspectos aunque cabe recalcar que no se ha curado por completo. Espero que estas investigaciones sigan en pie, para así en un futuro poder ayudar a muchas otras personas con sus enfermedades.
El artículo me parece excelente, los diverso puntos tratados se explican y entienden perfectamente. El tema es muy atrayente, pues yo nunca había escuchado sobre la medicina hiperpersonalizada o la enfermedad de Batten, lo recalco,es un contenido que me deja satisfecha con la explicación dada.
Este articulo desde mi punto de vista me parece muy interesante pero a la vez intrigante ya que tarta de un tema un poco desconocido hasta ahora , como lo es la creación de un medicamento personalizado es decir hecho específicamente para una sola persona con el fin de curar enfermedades que nos son tan comunes como otras.En la actualidad se están realizando varias investigaciones para determinar si esta practica es beneficiosa o perjudicial e incluso si conlleva alguna consecuencia.Ademas me parece que esta es una gran opción para ayudar a las personas a superar la enfermedad que les hayan diagnosticado., como es sel caso de Mila, a quien le han diagnosticado Batten una rara mutación de los genes, lleva utilizando un medicamento solo para ella por casi u año y se puede evidenciar una mejora peor no se puede decir que ya a superado su enfermedad.Espero que las investigaciones tenga resultados positivos para que se pueda aplicar esta técnica en el futuro y así ayudar a otras personas a vencer sus enfermedades.
Este articulo se me hizo muy interesante, ya que explica de una manera clara sobre la medicina hipersonalizada, pero antes de eso explica a grandes rasgos sobre la enfermedad, en donde se encuentra la alteración , así que se me hace muy complejo y claro , y sobre todo muy interesante ya que es la primera vez que leo algo sobre esta medicina , y el contenido se me hizo muy completo ya que sin tanto texto me dejo en claro cada uno de los puntos abordados en el mismo.
[…] milagro de Mila o la medicina hiperpesonalizada. Ya os contamos en este post todo el caso de Mila. Solo recordaros que se trata de la primera vez que se diseña un medicamento […]
Esto es una farsa para los humanos de a pie. Será tan costoso que tendremos que poner nuestra vidas a crédito para poder pagar una estúpida pastilla.
Está claro que no interesa la vida humana personalizada, solo nuestra cartera.
Me meo cuando leo estas noticias esperazadoras y traicioneras de doble filo.
Noticias para darse publicidad únicamente y tener sus dos minutos de gloria.
Que les den, no me trago nada, lo único en lo que creo es «tanto tienes, tanto vales» y sino, ahí estamos obteniendo tratamientos a granel, como si fuéramos ganado y ellos veterinarios en vez de médicos tratando a humanos.